Dr. Eric Olson, UT Southwestern Medical Center, overviewed recent work from his laboratory on a family of regulatory RNAs known as microRNAs (miRs) in cardiac and muscle diseases. Pathologic remodeling of the heart involves myocyte hypertrophy, apoptosis and fibrosis, a shift in cardiac metabolism, and fetal cardiac gene activation. The hallmark of fetal gene activation is a switch between the α- and β-myosin genes, which is a marker for many forms of cardiac disease. The process of cardiac remodeling includes changes in expression of protein coding genes. Dr. Olson’s laboratory investigated the question of whether there are changes in miR genes associated with heart disease.
There are as many as 1,000 human miR genes. The regulatory potential of miRs comes from their base-pairing with the 3-prime (3') untranslated regions of target messenger RNAs (mRNAs). The miR can either promote the cleavage or translational repression of mRNA. At least one-third of all human genes are subject to regulation by a miR-based mechanism. MiRs are secreted from cells in the heart and circulate. Numerous studies have shown that miRs in the circulation are diagnostic for a number of diseases.
Dr. Olson’s laboratory used microarray technology to profile the spectrum of miRs expressed in normal hearts versus diseased hearts (mouse model of hypertrophy and fibrosis). The experiment showed that each form of heart disease was characterized by a specific signature pattern of miRs, some of them upregulated and some downregulated. Other studies showed that miRs that are dysregulated in rodent models are also dysregulated in humans. Some changes in the miRs are pathogenic, while others are protective. For example, miR-195 induces myocyte loss and cardiac failure in a mouse model; inhibiting miR-195 promotes cardiac survival. Injury to the heart represses miR-29, leading to upregulation of extracellular matrix proteins and fibrosis.
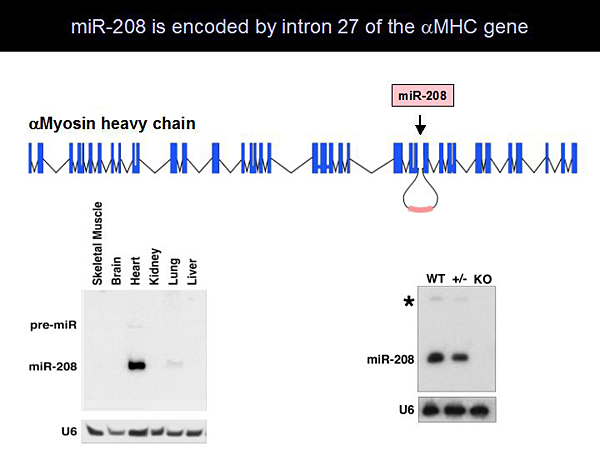
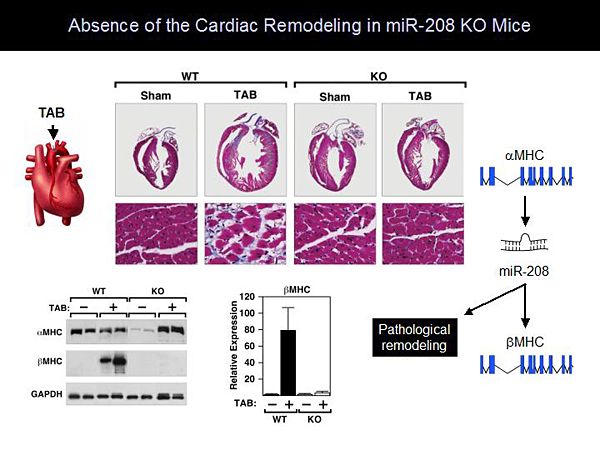
Dr. Olson and colleagues found that miR-208 is encoded by intron 27 of the amyosin heavy chain (aMHC) and is specifically expressed in the heart (Figure 1). Generation of miR-208 knockout (KO) mice resulted in mice with a normal phenotype. Even when stressed with pressure overload, these mice had no cardiac remodeling. Molecular analysis showed that miR-208 KO mice cannot upregulate bMHC at the protein or RNA level. These observations led to development of a model showing that aMHC regulates cardiac contractility and encodes miR-208, which is required for pathologic remodeling (Figure 2). Further studies showed that injection of anti-miR-208 inhibits the expression of miR-208 in the heart by at least 90%. This inhibition is sustained for at least 60 days following a single injection. Treatment with anti-miR-208 prevented hypertrophy and fibrosis in mice after pressure overload stress.
|
Figure 1. miR-208 is Encoded by Intron 27 of the aMHC Gene.
【Click to enlarge】 |
|
|
|
Figure 2. Absence of Cardiac Remodeling in miR-208 KO Mice.
【Click to enlarge】 |
|
|
|
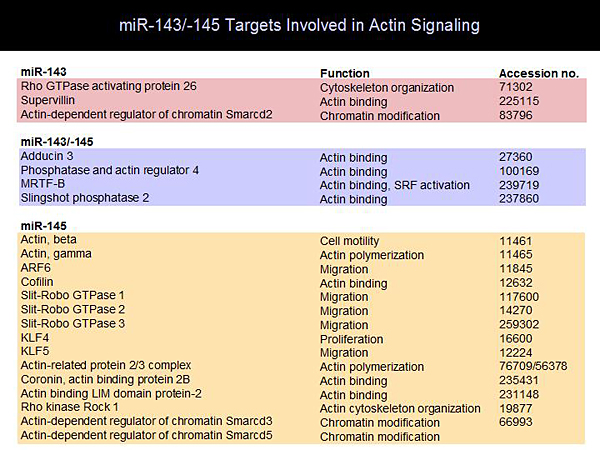
Figure 3. miR-143/-145 Targets Involved in Actin Signaling.
【Click to enlarge】 |
|
|
|
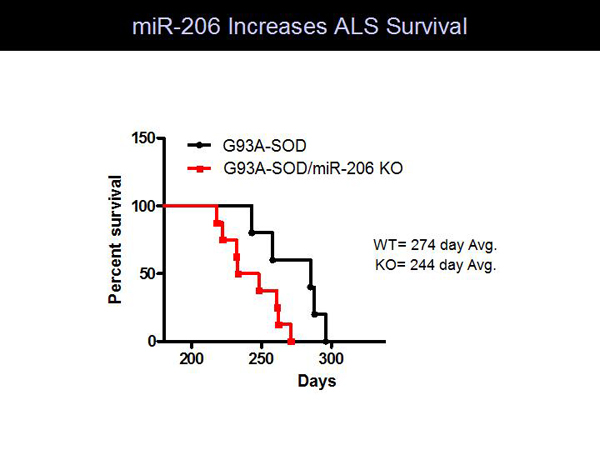
Figure 4. miR-206 Increases ALS Survival.
【Click to enlarge】 |
|
|
|
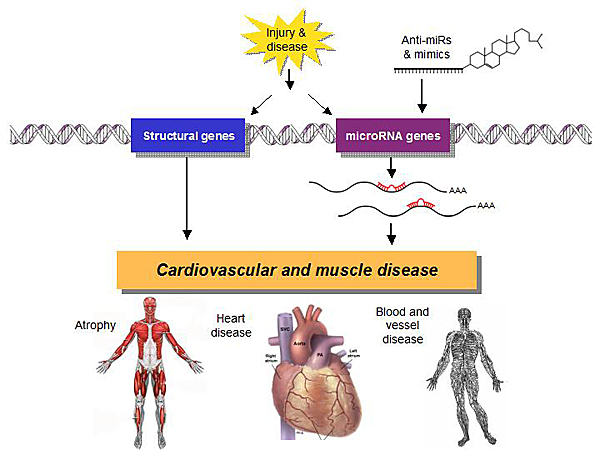
Figure 5. miRs in Cardiovascular and Muscle Disease.
【Click to enlarge】 |
|
|
Two other myosin heavy chains were found to encode miR-499 (Myh7b) and miR-208b (Myh7). These myosin-encoded miRs (myomiRs) function within an elaborate network to regulate stress responsiveness, thyroid hormone signaling, and other cardiac and skeletal muscle functions. The myomiRs work through a collection of proteins that repress transcription of the gene programs involved in stress responsiveness in slow myosin expression in the heart.
Activation of smooth muscle proliferation is the basis of many vascular disorders, such as restenosis and hypertension. The myocardin transcription factors (myocardin, MRTFs) are coactivators in this process. A microarray screen for MRTF-regulated miRs showed 9 miRs that were upregulated in response to MRTF versus control. MiR-143 and miR-145 were highly upregulated and were found to be encoded by the same gene. Mice with miR-143, miR-145, or both knocked out were normal, except for significantly reduced blood pressure, which probably is a reflection of relaxed vascular tone. Carotid artery ligation in miR-143/145 KO mice did not cause proliferation of the smooth muscle cells as would normally be expected. Analysis of the smooth muscle cells of these mice demonstrated that the actin could not organize into stress fibers as they do in wild-type (WT) mice. Many components of actin signaling are activated by miR-143 and miR-145) (Figure 3). These studies show that myocardin transcription factors are at the center of actin signaling and smooth muscle phenotypes.
Profiling of muscle miRs in amyotrophic lateral sclerosis (ALS) mice revealed that miR-206 was highly upregulated during disease onset. MiR-206 is expressed only in skeletal muscle, with its highest expression in slow muscle, which is resistant to ALS pathogenesis. Severing the motor nerve of a mouse caused a 50-fold increase in miR-206. Mouse studies showed that loss of miR-206 accelerates ALS pathogenesis and decreases survival compared to WT mice (Figure 4).
MiR-206 appears to mediate neuromuscular cross-talk. Muscle injury or denervation causes increased expression of miR-206, which generates a signal that promotes reinnervation and nerve muscle interactions. Mice lacking miR-206 have accelerated muscle atrophy following denervation and ALS.
MiRs are emerging as critical regulators of disease processes with direct relevance to human biology. Many miRs are up- and down-regulated in heart disease and other diseases (Figure 5). Studying the downstream targets of these miRs opens new dimensions in the understanding of the biology of disease and enables manipulation of miRs for therapeutic purposes.
|