|
|
Plenary Session 4 |
| |
| Cardiomyopathy UP to DATE: Molecular Basis of Cardiomyopathies – Recent Advances |
|
| |
|
| Diagnosis of ARVC |
| William J. McKenna, MD |
| Institute of Cardiovascular Science, University College London, UK |
| |
William J. McKenna, MD, University College London, focused his presentation on the diagnosis of arrhythmogenic right ventricular cardiomyopathy (ARVC). ARVC is characterized by structural, functional, and electrophysiological abnormalities secondary to fibro-fatty replacement in the right ventricular (RV) myocardium.
The gold standard of diagnosis is myocytes in various stages of cell death being replaced by fibrous tissue, with normal myocytes elsewhere. ARVC presents in three phases:
 |
Concealed: sporadic ventricular ectopic beats, subtle ECG/morphologic abnormalities, possible sudden death |
|
Overt: arrhythmia symptoms or sustained ventricular tachycardia (VT), diffuse RV/LV structural abnormalities, possible sudden death; |
|
Advanced: increased dilatation, decreased contractility of RV/LV, heart failure symptoms, sustained VT. |
Most patients who carry genes for ARVC are in the concealed phase, and if they survive, move into the advanced phase.
|
Figure 1. ARVC Diagnostic Criteria 2010.
【Click to enlarge】 |
|
|
|
Figure 2. Variable ECGs in Individual with Desmoplakin Mutation.
【Click to enlarge】 |
|
|
|
Figure 3. Disease-Causing Mutations in Desmosomal Proteins.
【Click to enlarge】 |
|
|
|
Figure 4. Arrhythmogenic Cardiomyopathy.
【Click to enlarge】 |
|
|
No single diagnostic test is definitive for ARVC. The features are non-specific, difficult to interpret, and subtle in the concealed phase. In 1994, a task force published a set of criteria for ARVC with high specificity but low sensitivity. The criteria were modified in 2010, incorporating more detail in terms of definition, a broader scoring system, recognition that LV involvement occurs, and severity classification based on numbers (Figure 1).
T wave inversion in RV leads V1-V3 and presence of epsilon waves as markers of slow conduction are considered characteristic of ARVC. Figure 2 shows ECGs of an individual with a desmoplakin gene mutation. The echocardiogram and cardiac magnetic resonance (CMR) are normal but the ECG is variable over time. A systematic analysis found similar variability in 20% of a study cohort, particularly with T wave inversion in the precordial leads. This suggests that electrical abnormality occurs before visible structural abnormalities. The first ARVC gene identified was plakoglobin, a desmosomal protein that links across the cell membrane to the junction and binds to desmoplakin and then to the intermediate filament (Figure 3). Desmosomal protein mutations account for 40%-60% of cases. These genes equally involve the LV. Most centers do not perform biopsy because of safety issues and the patchy nature of disease. A small study evaluated immunohistochemistry of desmosomal and adherin proteins. ARVC patients had normal adherin staining but plakoglobin was altered regardless of the underlying disease-causing gene, suggesting that plakoglobin may represent a final common pathway of disease. Dr. McKenna concluded that ARVC is a small component of desmosomal disease (Figure 4). Desmosomal mutations are not rare and occurred in about 15% of Dr. McKenna’s series. To what extent they are important in determining unexplained electrocardiographic abnormalities and arrhythmias remains to be determined. “The diagnostic criteria and approach to this in the genetic era should be leading us into a broader and newer understanding of this condition,” Dr. McKenna said.
|
| |
| Page Top |
|
| |
|
| Cardiomyopathy UP to DATE: Molecular Basis of Cardiomyopathies – Recent Advances |
| Akinori Kimura |
| Tokyo Medical and Dental University |
| |
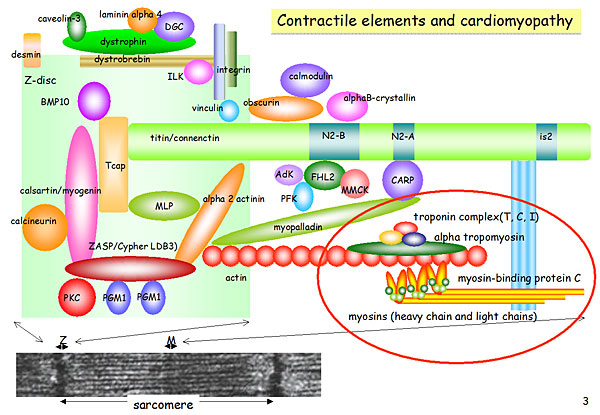
Dr. Akinori Kimura, Tokyo Medical and Dental University, discussed recent advances in identification and characterization of mutations associated with cardiomyopathies. The five clinical phenotypes of hereditary cardiomyopathy are: Hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), restrictive cardiomyopathy (RCM), arrhythmogenic right ventricular cardiomyopathy (ARVC), left ventricular non-compaction cardiomyopathy (LVNC), and storage disease with cardiac hypertrophy. Multiple gene mutations have been found for each type of cardiomyopathy in the contractile, Z-disc, I-band, sarcolemmal, and nuclear components.
|
Figure 1. Contractile elements and cardiomyopathy.
【Click to enlarge】 |
|
|
|
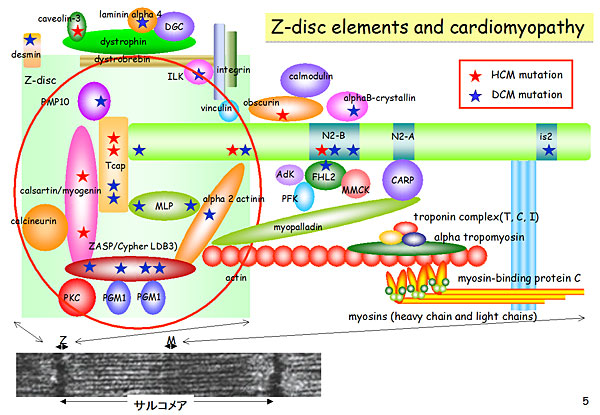
Figure 2. Z-disc mutations in HCM and DCM.
【Click to enlarge】 |
|
|
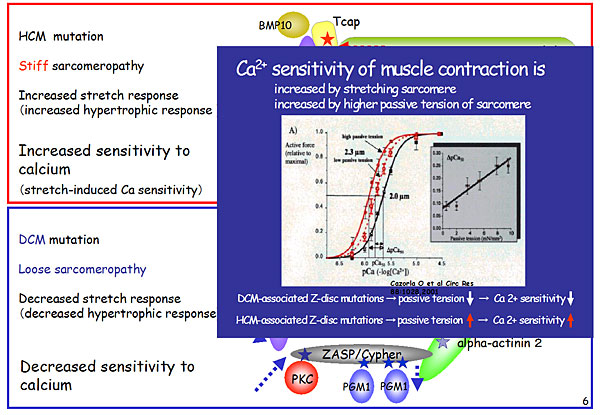
In the contractile component, troponin T mutations alter the calcium sensitivity of contraction (Figure 1). These mutations increase calcium sensitivity in HCM (del E160) and decrease calcium sensitivity in DCM (del K210). Several mutations have been identified in the Z-disc component, including functional mutations in HCM and DCM (Figure 2). In HCM, Tcap mutations lead to stiff sarcomeropathy and increased stretch response, resulting in increased calcium sensitivity of muscle contraction. In contrast, DCM mutations cause loose sarcomeropathy and decreased stretch response, leading to decreased calcium sensitivity.
A novel cardiomyopathy mutation involving the cardiac ankyrin repeat protein (CARP) has been identified in the I-band/nuclear component (Figure 3). Normally expressed in fetal cardiomyocytes, CARP expression in adults is induced by cardiomyocyte damage. CARP binds to the N2A region of tinin/connectin, the N-terminus of myopalladin, and desmin. It is a transcription co-factor that negatively regulates expression of cardiac remodeling genes, including MYL2, TNNC1, and ANP. Kimura’s group identified three CARP mutations in HCM patients—Pro52Ala, Thr123Met, and Ile280Val. When introduced to rat cardiomyocytes, mutant CARP was distributed to peri-nuclear or nuclear regions in the absence of stretch, leading to a constitutive stretch response.
|
Figure 3. I-band/nuclear element CARP and cardiomyopathy
【Click to enlarge】 |
|
|
|
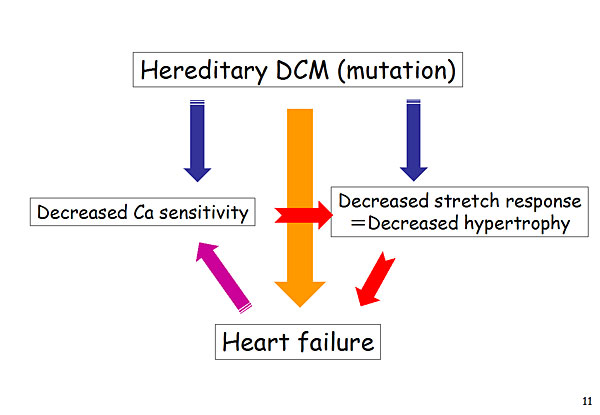
Figure 4. Hereditary DCM (mutation).
【Click to enlarge】 |
|
|
These studies show that hereditary DCM can be caused by several mutations, leading to decreased calcium sensitivity and decreased stretch response, resulting in heart failure (Figure 4). Further studies showed that treatment with the calcium sensitizer, SCH00013, prolonged survival in DCM phenotype mice. Echocardiography and histology studies showed that SCH00013 improved cardiac function and prevented expression of the pathological DCM phenotype (cardiac fibrosis) in the DCM mice. Cardiac remodeling also was attenuated in the SCH00013-treated DCM mice compared to non-treated DCM mice.
In conclusion, HCM-associated mutations cause increased calcium sensitivity, increased stiffness, and increased stretch response, leading to cardiomyocyte hypertrophy. DCM-associated mutations cause decreased calcium sensitivity, decreased stiffness, and decreased stretch response, leading to less sensitivity to hypertrophy. Development of hereditary cardiomyopathy could be prevented by modulation of calcium sensitivity.
|
| |
| Page Top |
|
| |
|
| Cardiomyopathy: A Disease of Protein Processing? |
| Peter F. Liu |
| University of Toronto, Canadian Institutes of Health Research, Canada |
| |
Dr. Peter F. Liu, University of Toronto, discussed mutations leading to abnormal protein processing that can result in DCM. A 2002 study reported that actin mutations and cytoskeleton disruptions can lead to DCM. Gelsolin, a protein located in the focal adhesion kinase, is elevated in heart failure and cardiomyopathy. When activated in the presence of cell surface stresses via calcium and PI3 kinase signaling, gelsolin severs actin filaments, initiating the remodeling process.
|
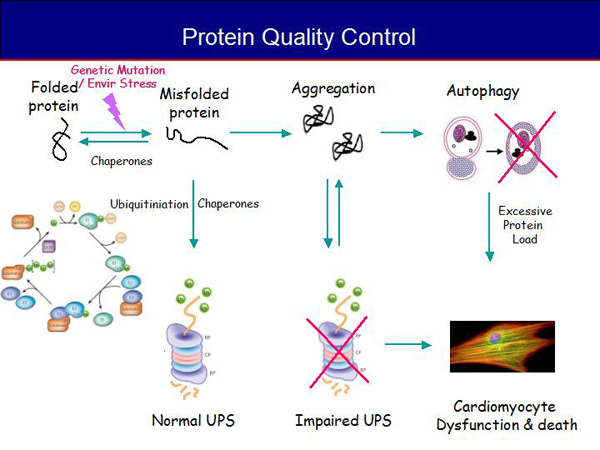
Figure 1. Protein Quality Control.
【Click to enlarge】 |
|
|
|
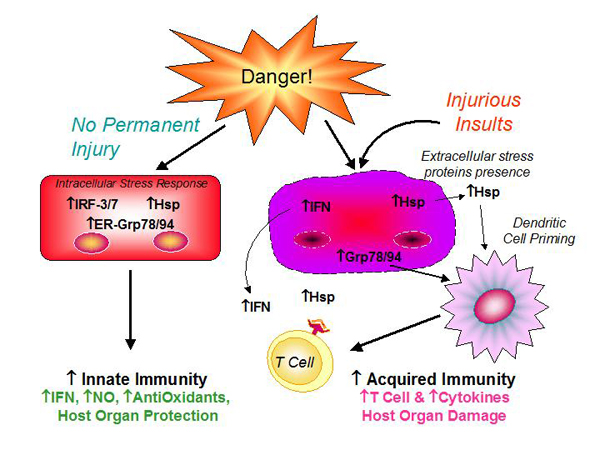
Figure 2. Activation of Stress Proteins and Immune Response.
【Click to enlarge】 |
|
|
Dr. Liu’s group investigated the role of gelsolin in the remodeling process using a myocardial infarction (MI) model. In gelsolin knockout mice with induced MI, dilation is attenuated and the remodeling process is decreased. Myocyte hypertrophy and cross-sectional area are decreased, and survival is increased (p <0.001). Some of these changes are related to decreased apoptosis and decreased fibrosis.
Another study reported that actin fragmentation by gelsolin activates proapoptotic programs, leading to direct DNA fragmentation and apoptosis. This process interacts with mitochondrial metabolism, further increasing oxidative stress. Thus, the fragmentation process leads to significant remodeling.
Mutations that lead to desmin changes may activate abnormal protein deposition inside the cell. A 2004 study found that mutation of the a-B-crystallin heat shock protein, which has chaperone functions for many cytoskeleton proteins, leads to desmin abnormalities and cardiomyopathy. The mutation causes extensive amyloid deposition in the myocytes in HCM and DCM. In transgenic mice with the mutation, exercise causes significantly decreased apoptosis, decreased caspase activation, and preserved cardiac function.
Figure 1 summarizes these processes. Terminally misfolded proteins are processed by the ubiquitin proteasome system (UPS) for degradation. If the UPS is overloaded, misfolded proteins may aggregate as amyloid fibrils, leading to autophagy and further impairment of the UPS. The authophagy system can be overwhelmed, leading to excessive deposition and myocyte dysfunction and death.
Proteomic and gene expression analysis revealed two protein subtypes consistently present in cardiomyopathies. The endoplasmic reticulum stress response proteins are associated with activation of apoptotic mechanisms via the caspases. Excessive accumulation of the proteins activates the unfolded protein response, leading to changes in transcription of genes important in remodeling, growth, and dilation. These processes can lead to activation of the immune response (Figure 2).
Other studies showed that: Pressure overload does not induce cardiac hypertrophy in Hace1-/- knockout mice; cathepsin-L deficiency impairs myocyte protein turnover, increasing aggregate formation and myocyte filament disorganization; cathepsin-L deficiency worsens ventricular remodeling and heart failure and systolic and diastolic function in response to pressure overload.
Dr. Liu concluded that therapies may involve reduction of abnormal protein accumulation, removal of abnormal stress, or improvement of UPS-autophagy-lysosomal removal mechanisms through exercise, increased blood flow, or new molecules.
|
| |
| Page Top |
|
| |
|
| Specific Immunoadsorption Therapy Using a New Tryptophan Column in Patients with Refractory Heart Failure Due to Dilated Cardiomyopathy |
| Tsutomu Yoshikawa, MD |
| Keio University School of Medicine |
| |
Autoantibodies directed against the b1-adrenergic receptor (β1AR), muscarinic M2-receptor, myosin, Na-K-ATPase, and TnI play a role in the pathophysiology of DCM. Tsutomu Yoshikawa, MD, Keio University School of Medicine, discussed the use of specific immunoadsorption therapy to eliminate autoantibodies in patients with DCM.
|
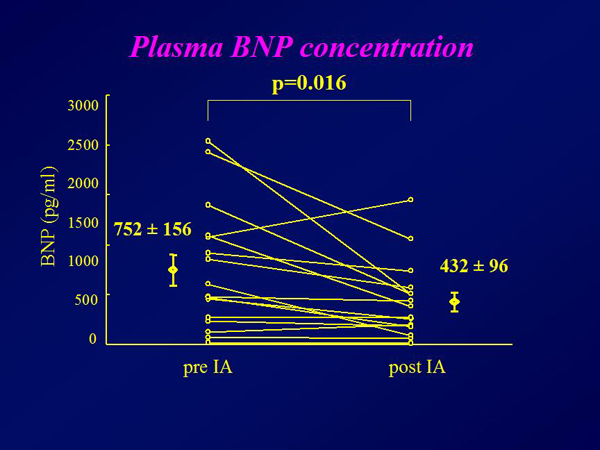
Figure 1. Plasma BNP concentration.
【Click to enlarge】 |
|
|
|
Figure 2. Left ventricular ejection fraction.
【Click to enlarge】 |
|
|
|
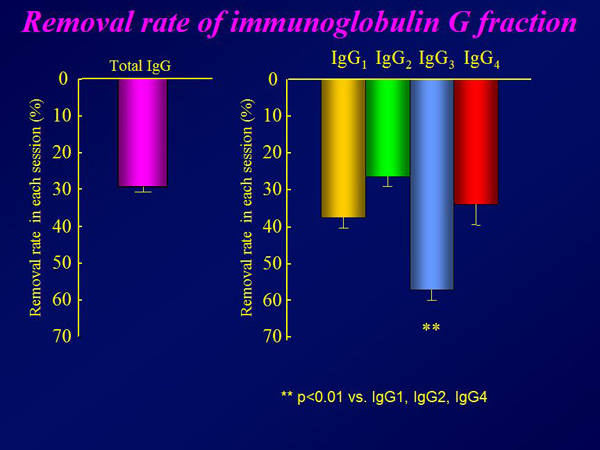
Figure 3. Removal rate of immunoglobulin G fraction.
【Click to enlarge】 |
|
|
|
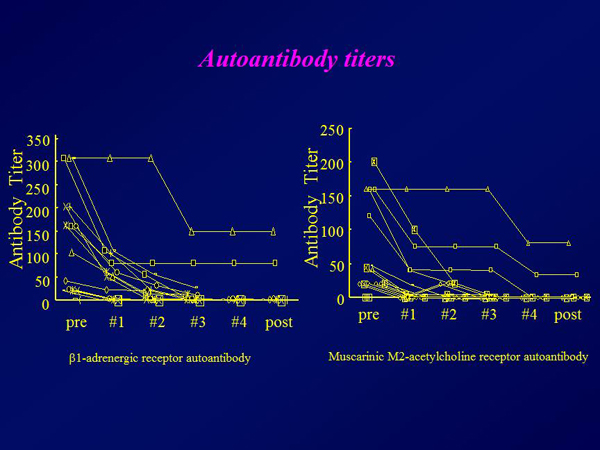
Figure 4. Autoantibody titers.
【Click to enlarge】 |
|
|
In a German study, DCM patients treated with immunoadsorption therapy using a column loaded anti-sheep IgG for 12 months had a significant increase in ejection fraction compared with the control group (p=0.0001). A study in patients with idiopathic DCM showed that β1AR antibody titers progressively decreased over 5 sessions of immunoadsorption therapy. Another study found that immunoadsorption increased ejection fraction in patients with cardiodepressant antibodies but not in patients without cardiodepressant antibodies (p<0.001 vs baseline; p<0.01 vs non-cardiodepressant group). Cardiodepressant antibodies belong to the IgG3 subclass. Elevated levels of IgG3 antibodies against α- and β-myosin heavy chains correlate with left ventricular dysfunction in patients with DCM.
Yoshikawa’s group studied immunoadsorption therapy using a high profile tryptophan column, which has low human antigenicity, high selectivity for IgG3, and does not require substitution of expensive immunoglobulin after the procedure. Three to five consecutive sets of immunoadsorption were performed in 16 patients with refractory heart failure caused by DCM. Approximately 2,000 mL of plasma were treated over 2 hours per procedure.
The plasma brain natriuretic peptide (BNP) concentration decreased from a mean 752 pg/mL to 432 pg/mL after completion of immunoadsorption therapy (p=0.016) (Figure 1). The mean ejection fraction increased from 18% at baseline to 21% (p=0.039) (Figure 2). The six-minute walk distance increased from 314 meters to 360 meters (p=0.01). Approximately 30% of the total IgG was removed in each procedure, with significantly greater removal of the IgG3 subclass than the other subclasses (Figure 3). Autoantibodies against the b1AR and muscarinic M2-receptor were completely eliminated in all but 2 patients, who had detectable levels of both autoantibodies after therapy (Figure 4). TNF-a and IL-6 levels remained the same after immunoadsorption therapy.
Based on these results, a randomized self and parallel group comparative trial of immunoadsorption plasma-apheresis in 40 patients with DCM was initiated. Dr. Yoshikawa concluded that cardiac-specific autoantibodies play a pivotal role in mediating the pathophysiology of DCM. Removal of these autoantibodies by immunoadsorption promises to be a therapeutic alternative in patients with refractory heart failure resulting from DCM. The use of the high profile tryptophan column, with its low antigenicity and high IgG3 specificity may be safe and effective in this context.
|
| |
| Page Top |
|
| |
|
| Current Status of Pathophysiological Survey of Dilated Cardiomyopathy in Japan: Report from the Research Committee of Idiopathic Cardiomyopathy |
| Hitonobu Tomoike, MD |
| National Cardiovascular Center, Suita, Japan |
| |
Hitonobu Tomoike, MD, National Cardiovascular Center, reported on data related to diagnostic and treatment standards from the Research Committee on Idiopathic Dilated Cardiomyopathy (DCM). The ongoing prospective, multicenter Cardiomyopathy Classification by Mortality and Morbidity (CCMM) study was initiated in 2005 to determine factors linked to prognosis in Japanese patients with cardiomyopathies and develop a new classification based on therapeutic evidence.
|
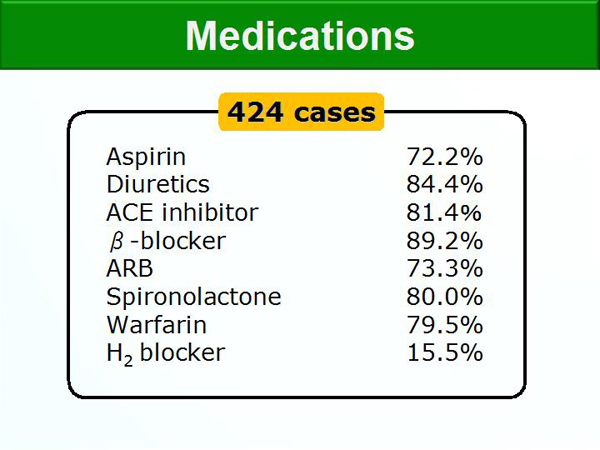
Figure 1. Medication Use.
【Click to enlarge】 |
|
|
|
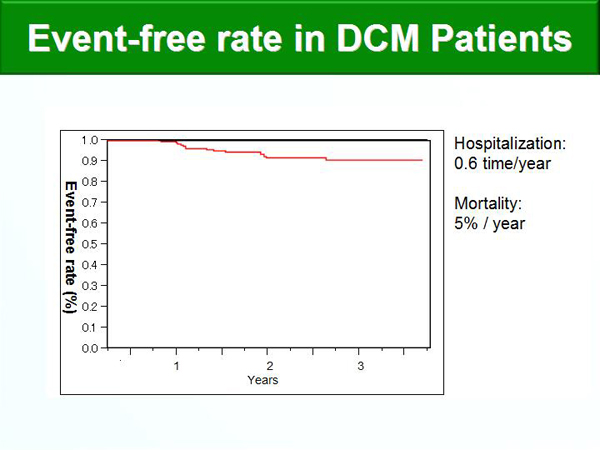
Figure 2. Event-Free Rate in DCM Patients.
【Click to enlarge】 |
|
|
|
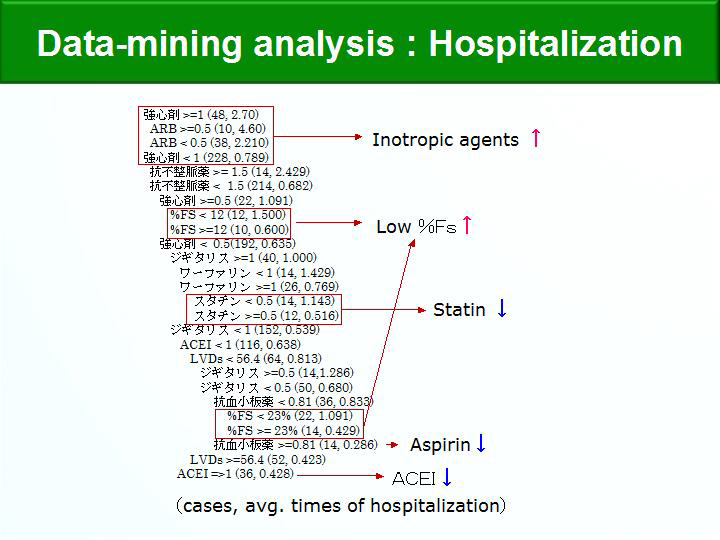
Figure 3. Data-Mining Analysis: Hospitalization.
【Click to enlarge】 |
|
|
|
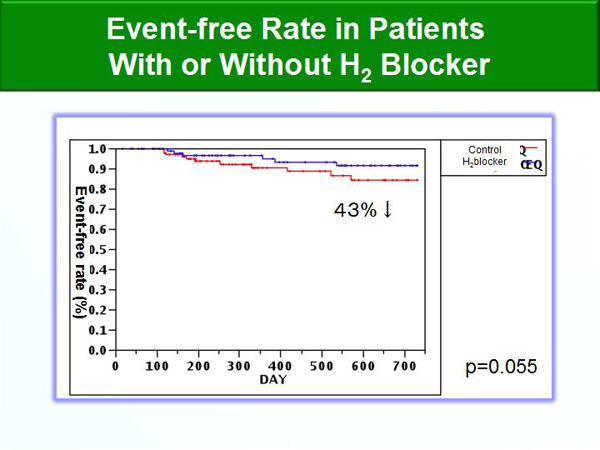
Figure 4. Event-Free Rate in Patients With and Without H2 Blocker.
【Click to enlarge】 |
|
|
|
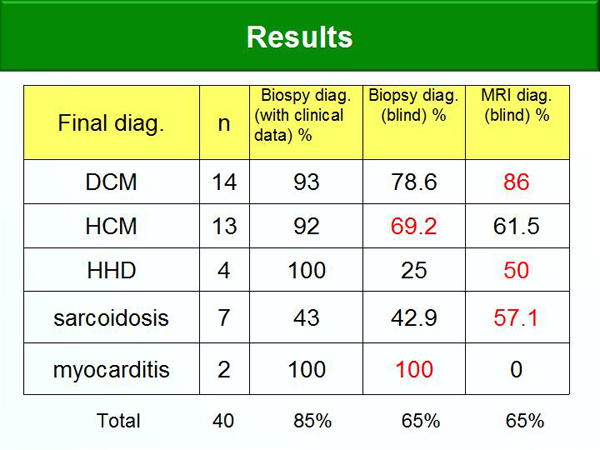
Figure 5. Comparison of MRI and Myocardial Biopsy Diagnosis Rates.
【Click to enlarge】 |
|
|
As of 2009, the CCMM study had accumulated data on 424 patients with DCM. Most of the patients had NYHA Class IIm and III disease. The average brain natriuretic peptide (BNP) level was 281.2 pg/mL, % fractional shortening (FS) was 16%, left ventricular diastolic dimension (LVDd) was 65.3 mm, LV systolic dimension (LVDs) was 54.9 mm, and left atrial dimension (LAd) was 42.8 mm.
The prevalence of impaired glucose tolerance and diabetes mellitus ranged from about 75% in patients with NYHA Class IIs, 85% in patients with NYHA Class IIm, and >90% in those with NYHA Class III versus about 20% in the general population. Figure 1 shows the medications the patients were receiving.
The annual hospitalization rate was 0.6, the mortality rate was 5% per year, and the 5-year mortality rate was 75% (Figure 2). Data-mining analysis was used to identify prognostic factors. Factors associated with increased re-hospitalization were low %FS and use of inotropic agents; factors associated with decreased re-hospitalization were use of statins, anti-platelet agents, and ACE inhibitors (Figure 3). Factors associated with decreased cardiovascular deaths were low BNP level and use of beta-blockers.
A subgroup analysis of the effect of histamine H2 receptor blockade was conducted. A mathematical model that predicts the level of repair, called the propensity score, was used to match the patients with minimal bias. Patients receiving H2 blockers (N=66) had 43% fewer events than those not receiving beta-blockers (N=132) (Figure 4).
A second subgroup analysis compared cardiac MRI and myocardial biopsy for DCM diagnosis in 14 patients. The diagnosis rate was highest with biopsy with clinical data (93%) but the diagnosis rate with MRI without clinical data (86%) was higher than that of biopsy without clinical data (78.6%) (Figure 5).
As previously reported for CHF, H2 receptor blocker therapy is effective in patients with DCM. Dr. Tomoike concluded that a nationwide registry of DCM patients is necessary to understand the profile of DCM in the Japanese and to explore improved diagnosis and treatment approaches for this intractable disease.
|
| |
| Page Top |
|
| |
|
|